Ausgangslage
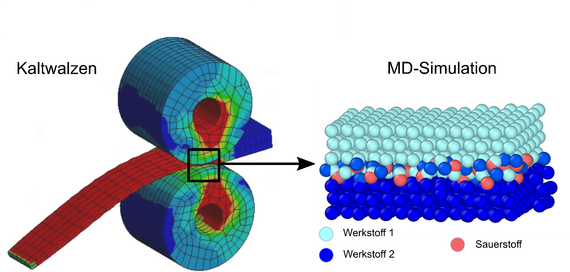
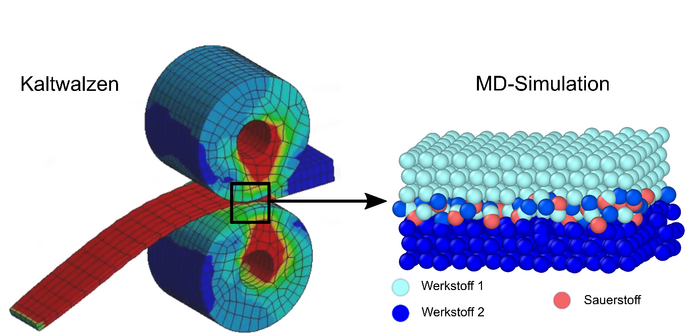
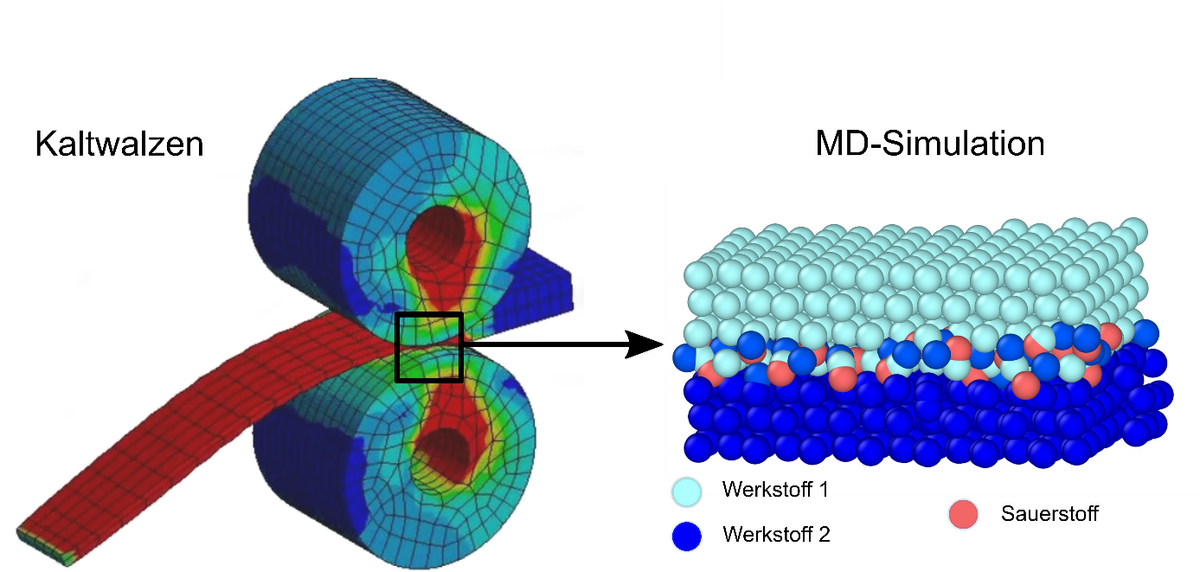
Beim Fügen bestimmt eine Vielzahl physikalischer und chemischer Prozesse die Festigkeit der entstehenden Verbindung. Eine analytische Vorhersage des Fügeprozesses ist damit nicht mehr ad hoc möglich, da kleine Variationen in der chemischen Zusammensetzung den Oberflächenzustand deutlich verändern und damit bereits zu großen Veränderungen des Fügeprozesses führen können.
Beim Fügen durch Kaltwalzplattieren spielt die Oxidation der Oberflächen der Fügepartner eine wichtige Rolle. Dies ist vor allem für Leichtbauwerkstoffe wie Aluminiumlegierungen relevant, bei denen sich sogar unter Hochvakuumbedingungen binnen Sekunden eine Oxidschicht ausbildet. Selbst geringe Verunreinigungen der Oberflächen durch Oxidschichten beeinflussen die Adhäsionskräfte beim Walzplattieren und können schlimmstenfalls eine Verbindung der Oberflächen verhindern. In der Folge ist eine zusätzliche werkstoffspezifische Wärmebehandlung nötig, um stoffschlüssige Verbindungen zu generieren.
Ziele
Im Projekt soll der Einfluss der Oxidschichten auf die physikalischen Eigenschaften der Bindung der Fügepartner in der Kontaktzone am Beispiel des Walzplattierens mittels atomistischer Simulationen grundlegend untersucht werden. Durch die mikroskopische Modellierung mittels Molekulardynamik (MD) wird die große Anzahl physikalisch-chemischer Vorgänge der Makroskala auf die Wechselwirkung zwischen Atomen und Molekülen reduziert. Die Verwendung der MD-Methode zur Simulation des Sauerstoffeinflusses bietet sich an, da die Sauerstoffkonzentration im Modell durch Einstellung des Sauerstoffpartialdrucks beliebig variiert werden kann. Damit können die grundlegenden Abhängigkeiten der Fügequalität von der vom lokalen Desoxidationszustand simulativ bestimmt werden, was durch Experimente nicht zugänglich ist.
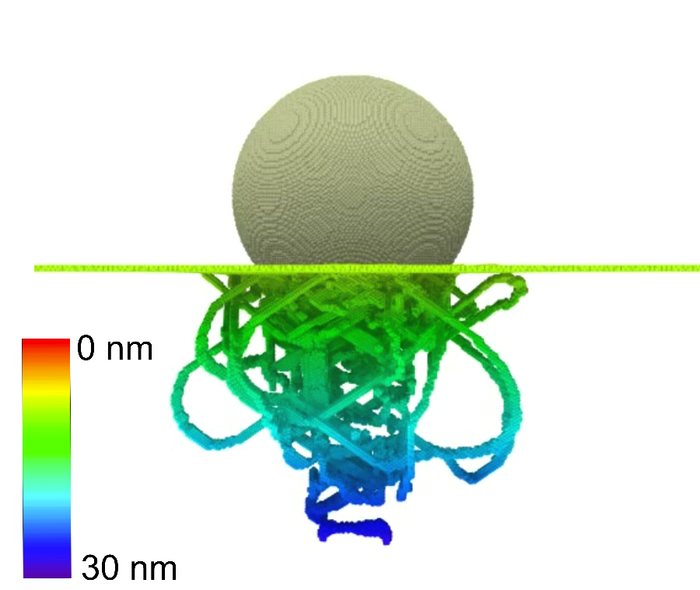
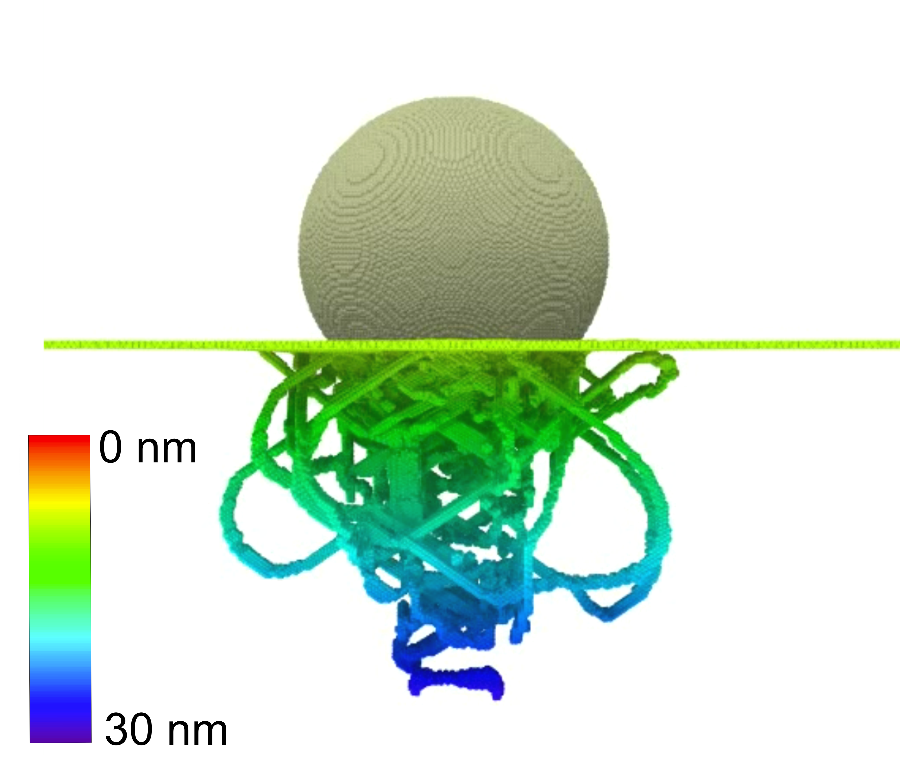
Es sollen Prozessbedingungen gefunden werden, die zu besonders hochwertigen, stoffschlüssigen Verbindungen führen. Die grundlegenden Abhängigkeiten der Verbundfestigkeit von Sauerstoffkonzentration, Temperatur sowie Oxidschichtdicke und -morphologie sollen für Grenzflächen untersucht werden. Die Härte der beteiligten Werkstoffe in der Umgebung der Grenzfläche als Kennzahl für die Qualität der Fügeverbindung wird mittels Nanoindentation in Teilprojekt C03 experimentell bestimmt und mit Simulationswerten verglichen.
Veröffentlichungen
Zeitschriftenbeiträge, begutachtet
-
(2023): Characterization of the tribologically relevant cover layers formed on copper in oxygen and oxygen-free conditions, Friction
DOI: 10.1007/s40544-022-0695-5 -
(2022): Molecular dynamics simulations of the machining of oxidized and deoxidized titanium work pieces, Results in Surfaces and Interfaces 9, p. 100085
DOI: 10.1016/j.rsurfi.2022.100085 -
(2022): Nanoindentation in alumina coated Al: Molecular dynamics simulations and experiments, Surface and Coatings Technology 437, p. 128342
DOI: 10.1016/j.surfcoat.2022.128342 -
(2022): Experimental and atomistic study of high speed collisions of gold nanoparticles with a gold substrate: Validation of interatomic potentials, Journal of Aerosol Science 159, p. 105846
DOI: 10.1016/j.jaerosci.2021.105846 -
(2021): Molecular dynamics simulation of nanoindentation in Al and Fe: On the influence of system characteristics, Applied Surface Science 551, p. 149221
DOI: 10.1016/j.apsusc.2021.149221 -
(2020): Molecular Dynamics Simulations of the Mechanical Behavior of Alumina Coated Aluminum Nanowires under Tension and Compression, RSC Advances 10 (2020), pp. 14353–14359
DOI: 10.1039/D0RA01206H
Dissertationen
-
(2023): Molecular dynamics simulations of plastic deformation in iron and aluminum, Dissertation, Technische Universität Clausthal
DOI: 10.21268/20230214-0
Verschiedenes
-
(2021): Molekulardynamik-Simulationen von Grenzflächenphänomenen in sauerstofffreier Atmosphäre (Vortrag), 4. Symposium Materialtechnik des CZM
-
(2021): Atomistic Study of the Influence of Oxide Shell Layers on the Material (Vortrag), 4. Symposium Materialtechnik des CZM
Teilprojektleiterin/Teilprojektleiter
Arnold-Sommerfeld-Straße 6
38678 Clausthal-Zellerfeld
Arnold-Sommerfeld-Straße 6
38678 Clausthal-Zellerfeld
Teilprojektbearbeiterin/Teilprojektbearbeiter
Arnold-Sommerfeld-Straße 6
38678 Clausthal-Zellerfeld
Arnold-Sommerfeld-Straße 6
38678 Clausthal-Zellerfeld

